Article 26a of the 2013 (Genetically Modified Organism) Decree establishes the simplified procedure for authorizations under fixed rules (VoV) for GMO applications in clinical studies whose environmental risks are well known based on experience and knowledge from previous environmental risk assessments.
Currently, the simplified procedure for a VoV is applicable for clinical studies involving the following categories of GMOs:
- genetically modified viral vectors derived from Adeno-associated dependoparvovirus A or B (AAV) without deleterious sequences (GMO Regulation Article 39a);
- human cells genetically modified with viral vectors derived from mouse gamma-retroviruses or human immunodeficiency virus (HIV), where there is no risk of replication-competent virus formation and where residual infectious retroviral or lentiviral particles are absent (GMO Regulation Article 39b);
- human cells genetically modified with viral vectors derived from human immunodeficiency virus 1 and pseudotyped with Vesicular stomatitis virus glycoprotein (VSV-G), where there is no risk of replication-competent virus formation and where residual infectious SIN lentiviral particles may be present in the medical product (GMO Regulation Section 39c);
- human cells genetically modified with viral vectors derived from Adeno-associated dependoparvovirus A or B without deleterious sequences (GMO Regulation Section 39d).
If the application meets the additional conditions below, a maximum target time of 28 or 56 days for processing the application applies.
AAV without harmful sequences
Article 40a of the 2013 (Genetically Modified Organism) Regulations sets out the requirements for a VoV permit application for the application of (Adeno-Associated Virus) in clinical trials. Of those requirements, only f, g and h are potentially relevant to distinguish between a processing within 28 or 56 days.
(f) Data regarding the molecular characterization of the genetically modified AAV vector
- 28 days if the identity of the vector genome (ITRs and intermediate sequences), or of the plasmids used for the manufacture in case of transient transfection, is verified by sequencing
- 56 days if molecular characterization was performed by other means
(g) Data on the possibility of formation of replication-competent AAV
- No need for differentiation
h) Data on the absence of infectious helper virus
- 28 days if no helper virus was used during production
- 28 days if wild-type helper virus was used
- 56 days if a GM helper virus was used
Ex vivo lentiviral/retroviral transduced cells withoout residual infectious retroviral or lentiviral particles
Article 40b of the 2013 (Genetically Modified Organism) Regulations sets out the requirements for a VoV permit application for the application of ex vivo lentiviral/retroviral transduced cells in clinical trials. Of those requirements, only f, h and i are potentially relevant to distinguish between a processing within 28 or 56 days.
(f) Molecular characterization data of the genetically modified retroviral or lentiviral vector
- 28 days if the identity of the vector genome (LTRs and intermediate sequences), or of the plasmids used for the manufacturing in case of (transient) transfection, is verified by sequencing
- 56 days if molecular characterization was performed by other means
h) Data on the absence of replication-competent retrovirus or replication-competent lentivirus
- 28 days for 3rd generation SIN lentiviral systems
- 56 days for other lentiviral systems and for retroviral systems
(i) Data on the absence of residual infectious retroviral or lentiviral particles in the cell product administered to the subject
- 28 days if the reduction ratio is calculated to be greater than 100 using the (Netherlands Commission on Genetic Modification) formula
- 56 days if the absence is otherwise calculated or demonstrated
Rationale
In recent years, much experience and knowledge has been gained with the application of gene therapy, especially when it comes to the safety of the "platform" used to make the actual therapeutics. These platforms consist of harmless viruses that are capable of entering a cell and making the necessary change in the cell's DNA without (too much) damage in the form of side effects. Because of this accumulated knowledge as a result of previous authorizations under the extended regular authorization procedure, the outcome of the environmental risk assessment (hereinafter: (environmental risk assessment)) for such viruses is nowadays known in advance, namely negligibly small. This means that for such (Genetically Modified Organism) applications there is no longer a need for an elaborate assessment to assess safety, which means that the decision period can be shortened. With the observation that the outcome of the ERA always arrives at "negligible risk," the need for prior public consultation has also become unnecessary. For such GMO applications, the simplified procedure is included in the 2013 GMO Decree through Article 3.26a.
Based on the aforementioned experiences, generic and broadly applicable ERAs have now been drawn up for some GMO applications of gene therapeutics. Within the scope of the ERA (the scope) there will always be negligible risk. Applicants do not have to perform an individual ERA for the GMO applications that fall within the scope of Article 3.26a, but can simply refer to the standardized ERA. This simplifies the preparation of the application and thus makes it ready faster. Because the assessment by the competent authority is simpler, the applicant also receives the license sooner and can start the clinical trial sooner.
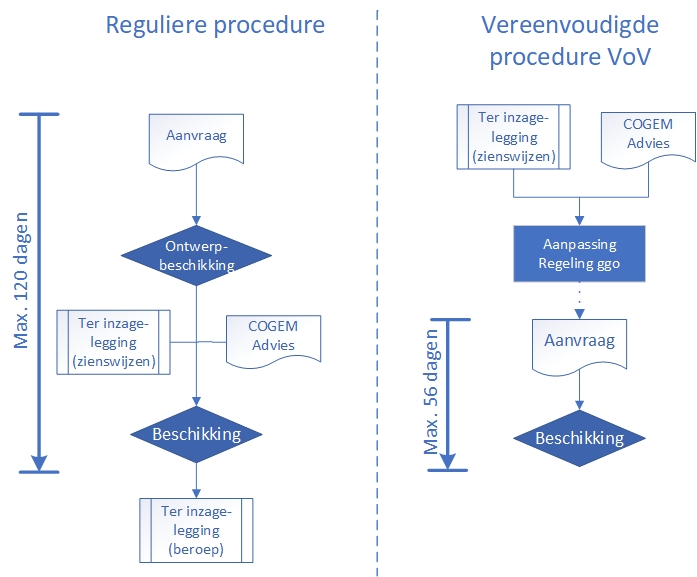
The figure below schematically shows the procedural distinction between the regular and the simplified procedure.
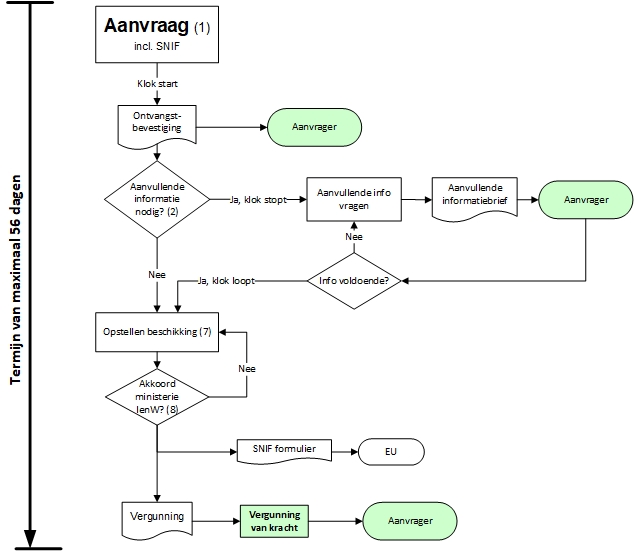
The procedural handling of the license application under fixed rules
The license application for gene therapy research must be submitted to the Ministry of (Ministry of Infrastructure and Water Management) (c/o (Office for Genetically Modified Organisms))(1). The procedure period starts the moment application is received. A confirmation of receipt is also sent. The procedure term can be stopped as soon as additional information is requested. The term will continue as soon as the requested additional information is received (2).
In addition to a permit application to the Ministry of IenW, the intended work must also be reported to the other member states of the European Community. The (Summary Notification Information Format) (Summary Notification Information Format) form available for this purpose can also be downloaded and should be submitted to Bureau GGO together with the permit application. Bureau GGO provides the form with a dossier number and forwards it to the European Commission after which it is published on the Internet.
A decision must be drawn up within eight weeks (7) and signed by the Ministry of Infrastructure and the Environment (8). The decision is sent to the researcher. After the permit is granted, it becomes effective immediately.
The figure below shows the procedural handling of the simplified procedure schematically.